
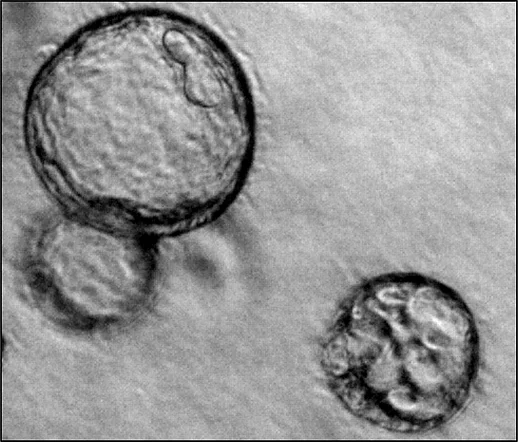
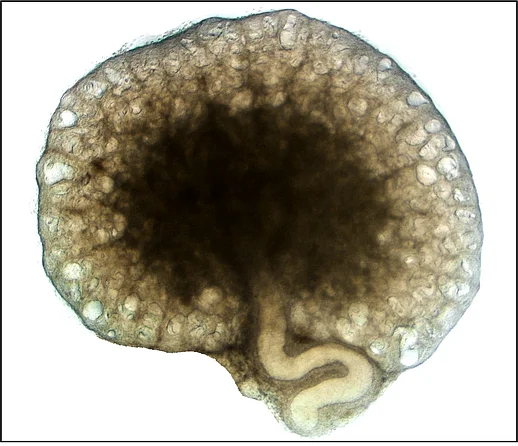

In our group we are mainly interested in the mechanisms of cyst growth in polycystic kidneys. There is a variety of polycystic kidney diseases that are often hereditary. The most frequent form is the autosomal dominant polycystic kidney disease ADPKD. It affects one in one thousand people and leads to the development of various fluid-filled cavities (cysts) in both kidneys. Cysts then grow continuously over time which causes compression of adjacent unaffected tissue which often results in loss of renal function. Cyst expansion can mainly be attributed to secretion of chloride and fluid by the cyst epithelium into the interior of the cyst. We investigate proteins but also environmental factors that may play a role in cyst secretion. Our long-term objective is to find strategies in order to inhibit cyst growth and therefore preserve renal function.
The Role of Hypoxia
Continuous cyst growth results in progressive hypoxia in polycystic kidneys which in particular leads to expression of the hypoxia-inducible factor HIF-1α in cyst-lining cells. We analyse the effects of HIF-1α on cyst expansion.
The Role of Anoctamins
Chloride secretion into the cyst lumen is mediated by apically expressed chloride channels. Next to the cAMP-dependent CFTR chloride channel also Ca2+-activated chloride channels are involved. Therefore, we investigate the impact of anoctamins, a new family of Ca2+-activated chloride channels, on cyst secretion.
The Role of Purinergic Receptors
Activation of Ca2+-activated chloride channels often follows stimulation of purinergic receptors (P2Y- and P2X receptors) by extracellular ATP. We study the relevance of purinergic receptors for secretion-dependent cyst growth.
Group leader

Scientist Post-doc

Scientist Post-doc University of Regensburg
Dr. rer. nat. Kathrin Skoczynski
E-Mail: kathrin.skoczynski(at)vkl.uni-regensburg.de
Technicians


Collaborations
- Prof. Dr. med. Karl Kunzelmann, Department of Physiology, University of Regensburg
- SFB 1350 Projekt A2, Universität Regensburg
- SFB 1350 Projekt B3, Universität Regensburg
- PhD Dorien J.M. Peters, Leids Universitair Medisch
Research group Kraus

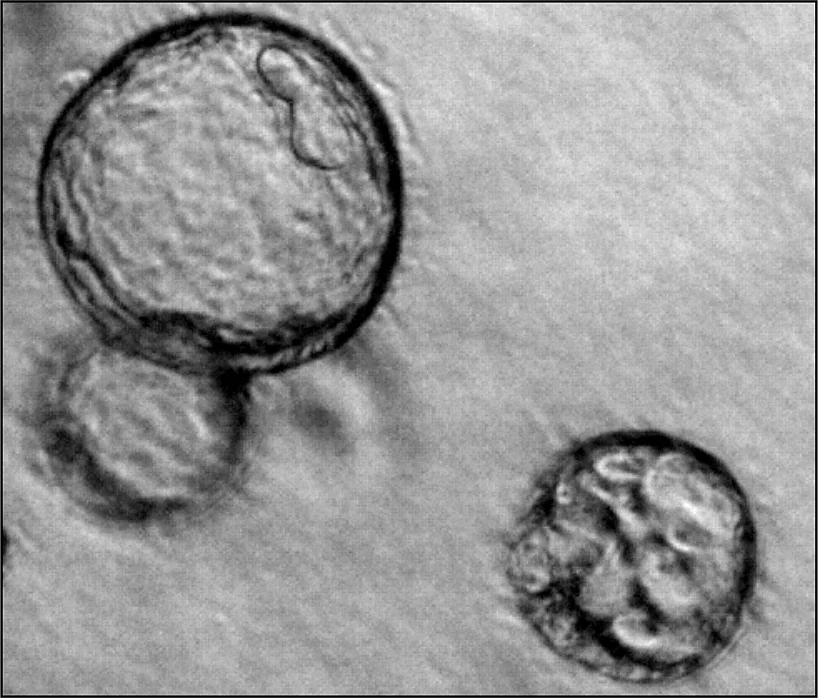
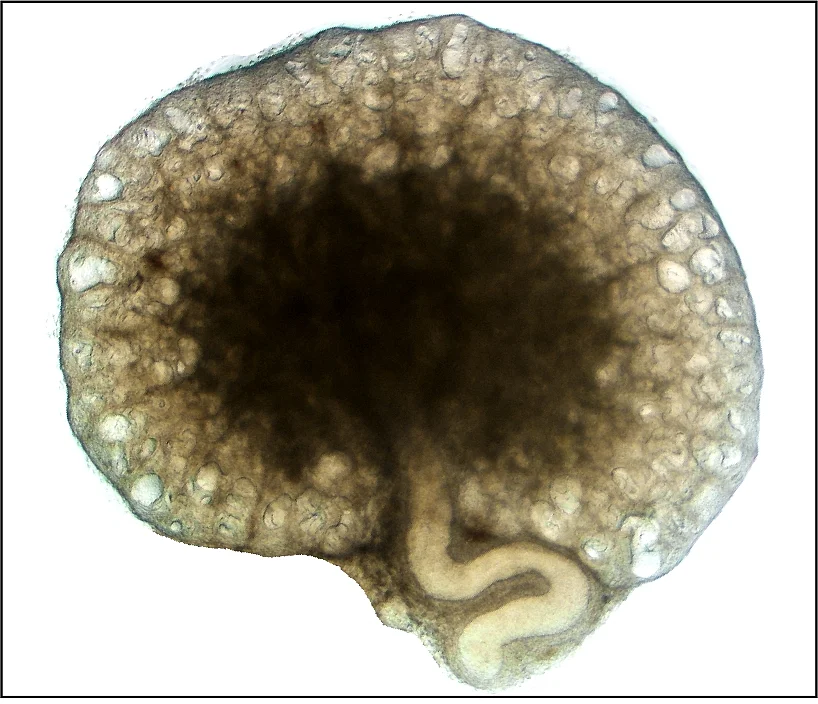

[Translate to English:] Mechanismen renalen Zystenwachstums
[Translate to English:]
In unserer Forschungsgruppe untersuchen wir die zugrunde liegenden Mechanismen des Zystenwachstums bei Zystennierenerkrankungen. Diese sind häufig genetisch bedingt; die am weitesten verbreitete Form ist die autosomal dominante polyzystische Nierenerkrankung (ADPKD). Etwa einer von tausend Menschen ist betroffen. Charakteristisch für ADPKD ist die fortschreitende Entwicklung zahlreicher flüssigkeitsgefüllter Hohlräume (Zysten) in beiden Nieren. Über Jahre hinweg wachsen diese Zysten kontinuierlich und verdrängen zunehmend gesundes Nierengewebe, was bei einem großen Teil der Patientinnen und Patienten letztlich zu einem Verlust der Nierenfunktion führt. Ein wesentlicher Treiber dieses Wachstums ist die Sekretion von Chlorid und Flüssigkeit durch das Zystenepithel in das Zystenlumen. Wir erforschen daher, welche Proteine und welche milieuabhängigen Faktoren diese Sekretionsprozesse beeinflussen. Langfristiges Ziel unserer Arbeit ist es, Strategien zu entwickeln, die das Zystenwachstum hemmen und die Nierenfunktion erhalten.
In diesem Zusammenhang testen wir zudem geeignete pharmakologische Substanzen, die das Zystenwachstum reduzieren könnten – in einem Spektrum von in vitro- bis in vivo-Modellen – mit dem klaren translationalen Ziel, vielversprechende Wirkstoffe in klinische Studien zu überführen.
Die Rolle von Hypoxie
Mit zunehmendem Zystenwachstum entsteht eine fortschreitende Hypoxie in der Zystenniere. Dies führt insbesondere zu einer verstärkten Expression des Hypoxie-induzierbaren Faktors HIF-1α im Zystenepithel. Wir untersuchen, wie diese veränderte HIF-1α-Expression das Zystenwachstum beeinflusst.
Die Rolle von Anoctaminen
Die Sekretion von Chlorid in das Zysteninnere erfolgt über luminal lokalisierte Chloridkanäle. Neben dem cAMP-abhängigen CFTR-Kanal tragen auch Ca²⁺-aktivierte Chloridkanäle wesentlich zu diesem Prozess bei. In diesem Kontext analysieren wir die Bedeutung der Anoctamine, einer neu identifizierten Familie Ca²⁺-aktivierter Chloridkanäle, für die sekretionsabhängige Zystenvergrößerung.
Die Rolle purinerger Rezeptoren
Die Aktivierung Ca²⁺-abhängiger Chloridkanäle wird häufig durch die Stimulation purinerger Rezeptoren (P2Y- und P2X-Rezeptoren) durch extrazelluläres ATP vermittelt. Wir erforschen, welchen Einfluss diese Rezeptoren auf das sekretionsgetriebene Zystenwachstum ausüben.

